В.В. Малиновская1, В.С. Сускова2, Е.Н. Выжлова1, А.Н. Шувалов1, С.И. Сусков3
1 Национальный исследовательский центр эпидемиологии и микробиологии имени почетного академика .Н. Ф. Гамалеи
2 Медицинский центр «Диагностика» (Москва)
3 Группа компаний «СМ-Клиника» (Москва)
Конец XX века ознаменовался достижениями в биологии и медицине, в том числе теоретической иммунологии, что позволило по-новому взглянуть на этиопатогенез ряда заболеваний. Речь, в частности, идет об изменениях, касающихся представлений о врожденном иммунитете, открытии системы интерферона, идентификации иммунорегуляторных клеток и субпопуляций лимфоцитов, синтезирующих оппозитно действующие цитокины, а также их роли в аутоиммунной патологии.
Ключевые слова: врожденный иммунитет, иммунорегуляторные клетки, апоптоз, интерферон, ВИФЕРОН
Последние годы представление о механизмах программируемой гибели клеток кардинально изменилось, что позволило выделить в самостоятельные формы апоптоз, некроз, аутофагию, митотическую катастрофу, клеточное старение и фагоцитоз, опосредованный презентацией на мембране «сигналов гибели» [1]. Особую значимость приобретает феномен апоптоза, открытый сравнительно недавно [2]. Установлено, что генетическая программа в клетках организма, обеспечивающая их жизненный цикл, при определенных физиологических или патологических условиях, в том числе вирусных инфекциях, запускает процесс апоптоза (программируемая гибель клеток) [3, 4].
Вирусы внутри зараженной клетки способны нарушать передачу сигналов рецепторами цитокинов и снижать апоптотическую активность, что требует поиска способов регуляции и коррекции апоптоза. Оценка апоптоза при клинико-иммунологическом обследовании пациентов с различными заболеваниями важна для обоснования методов коррекции апоптоза, в том числе с использованием интерфероновых препаратов.
Апоптоз – программируемая гибель клетки
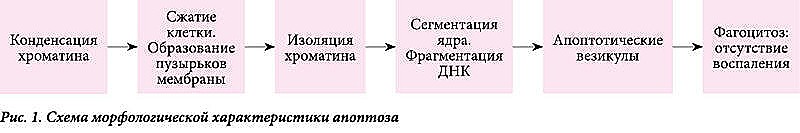
Нормальное развитие организма и функционирование иммунной системы поддерживаются балансом гомеостаза (соотношение между образующимися и отмирающими клетками). Апоптоз – необходимый инструмент морфогенеза и обеспечения нормального функционирования многоклеточных механизмов [5, 6]. Этот процесс регулируется программируемой (физиологической) гибелью клеток. В процессе апоптоза клеточные популяции очищаются от отработанных, нежелательных или поврежденных клеток. Первоочередными морфологическими признаками этого утонченного процесса служат конденсация хроматина и сжатие клетки. Далее клеточная мембрана формирует небольшие пузыри, и клетка начинает выталкивать свое содержимое внутрь везикул. В некоторые из них попадают части фрагментированного и конденсированного (пикнотического) ядра, что приводит к образованию апоптотических везикул, поглощаемых и разрушаемых макрофагами. Воспалительная реакция отсутствует, поскольку цитоплазматические ферменты и токсические метаболиты остаются окруженными мембраной клетки (рис. 1) [3].
![]()
Таким образом, структурная целостность биологических мембран предупреждает выход содержимого цитоплазмы, в том числе лизосомальных ферментов, во внеклеточную среду, что позволяет избежать структурных и функциональных дефектов ткани и воспаления при апоптозе. Апоптоз – генетически регулируемый процесс, для которого необходимы запас энергии и синтез определенных белков. Фрагментация клеток при апоптозе может стимулироваться набором сигналов, в том числе физиологическими стимулами (например, связывание антигена).
В качестве регуляторных сигналов рассматривают нарушение межклеточных контактов, удаление ростовых факторов, гипертермию или действие гранзимов. Общим внутриклеточным медиатором апоптоза может быть окислительный стресс (О2, Н2О2, NO, ОН-радикалы), вызывающий активацию нуклеаз, расщепляющих ДНК на фрагменты (рис. 2).
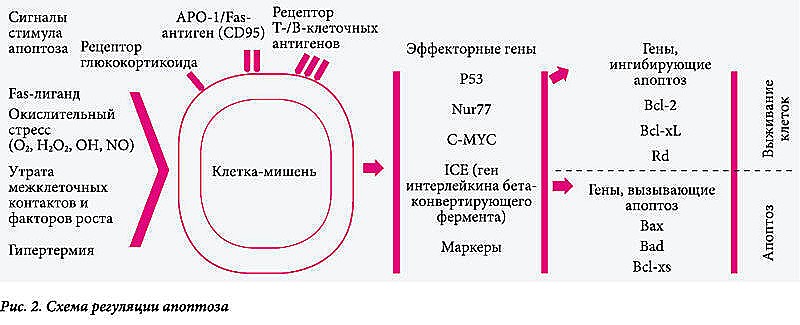
Структуры, участвующие во включении механизма апоптоза
Знания об апоптозе позволили сформулировать понятия позитивной и негативной активации клеток иммунной системы [7], необходимые для оценки их функционального состояния. Позитивной считается классическая активация лимфоцитов под влиянием специфических или неспецифических стимулов, способствующая реализации клеткой ее эффекторных функций (цитотоксичность, синтез иммуноглобулинов и цитокинов). Позитивная активация сопровождается повышением уровня экспрессии на Т- и В-лимфоцитах ряда активационных маркеров: CD25, CD40L, HLADR и др.
При негативной активации на лимфоцитах появляются активационный маркер CD95 (Fas/APO-l) и его лиганд FasL. Fas/APO-1 способен запускать процесс апоптоза после взаимодействия с лигандом FasL. Вследствие нарушения экспрессии Fas-рецепторов и других звеньев апоптоза развивается аутоиммунный лимфопролиферативный синдром, проявляющийся рядом заболеваний, характеризующихся доброкачественной лимфопролиферацией, гипериммуноглобулинемией и аутоиммунными расстройствами.
Одним из мембранных клеточных рецепторов, ответственных за контролируемый тканевой гомеостаз и иммунный ответ, является Fas-рецептор (CD95/APO-1) – белок с молекулярной массой 45 кD. Его функция связана со скоростью созревания и восстановления пула клеток. Кроме Fas-рецептора на поверхности многих гемопоэтических клеток присутствует еще один мембранный белок Fas-лиганд (FasL). Fas-лиганд имеет растворимую форму в виде белка с молекулярной массой 1,7 ? 104 D. Поверхностные молекулы типа CD95 (АРО-1 или Fas-антиген) – важные медиаторы апоптоза. Молекула CD95 принадлежит к рецепторным белкам семейства TNF (tumor necrosis factor – фактор некроза опухоли (ФНО))/NGF (nerve growth factor – фактор роста нервов). При активации АРО-1/Fas-лигандами клетка с рецептором CD95 (АPO-1/Fas-антиген) посылает сигнал апоптоза в ряд клеток, имеющих этот рецептор [3, 8].
Существуют специализированные рецепторы апоптоза, которые относятся к семейству TNF. Их общее обозначение DR (death receptors) (DR1-TNF, DR2 – Fas-рецептор, CD95, DR3, DR3 – DR6). Лигандами для TNFR1 служат TNF и лимфотоксин-альфа, для Fas-рецептора – мембранная молекула Fas-лиганд (FasL, CD178), для DR3 – DR3L.
Известно два основных рецептора, принимающих сигналы к развитию апоптоза, – Fas (CD95) и рецептор для TNF типа 1 (р55) TNFR1. Они имеют в цитоплазме «домен гибели», передающий внутрь клетки сигнал гибели (эффекторный ген IСЕ). Генерация внутриклеточных сигналов апоптоза прежде всего связана с белком р53, который экспрессируется при наличии поломок хромосом, разрывов ДНК и других генетических нарушений при разных сигналах, особенно ионизирующей радиации. Оставаясь в сморщенном виде, клетка утрачивает часть генетического (ядерного) материала при апоптозе [4].
Апоптоз сопровождается также активацией ряда генов. Одним из наиболее значимых является интерлейкин (ИЛ) бета-1-конвертирующий фермент (ICE). Для начальной фазы апоптоза также характерно повышение уровня экспрессии эффекторных генов p53, Nur77, c-MYC-белков. В процессе апоптоза помимо генов, вызывающих его (Bax, Bad, Bcl-xs), экспрессируются гены, ингибирующие апоптоз (Bcl-2 кодирует белок, предотвращающий апоптоз). Как следствие – кодирование процесса апоптоза и выживание клеток. Это особенно важно при патогенезе вирусных инфекций. Гены, вызывающие запуск апоптоза, приводят к его развитию (рис. 2).
Пути запуска апоптоза
Существует два механизма запуска гибели клетки – внутренний (митохондриальный) и рецепторный.
Митохондриальный апоптоз развивается при дефиците факторов, обеспечивающих выживаемость клеток (цитокинов и контактных сигналов от соседних клеток), а также под действием цитотоксических агентов (облучение, стероидные гормоны, цитостатики). В результате изменяется баланс митохондриальных факторов семейства Вcl-2 (проапоптотический и противоапоптотический). Через сформированные в мембране митохондрии в цитозоль выходит цитохром C, где он активизирует каспазу 9 путем связывания Apaf-1 с АТФ/дАТФ и прокаспазой 9. После этого дальнейший процесс апоптоза сопровождается образованием новых каспаз и разрушением клетки (рис. 3). В процесс вовлекаются инициаторные каспазы, мишенью которых служат исполнительные каспазы.
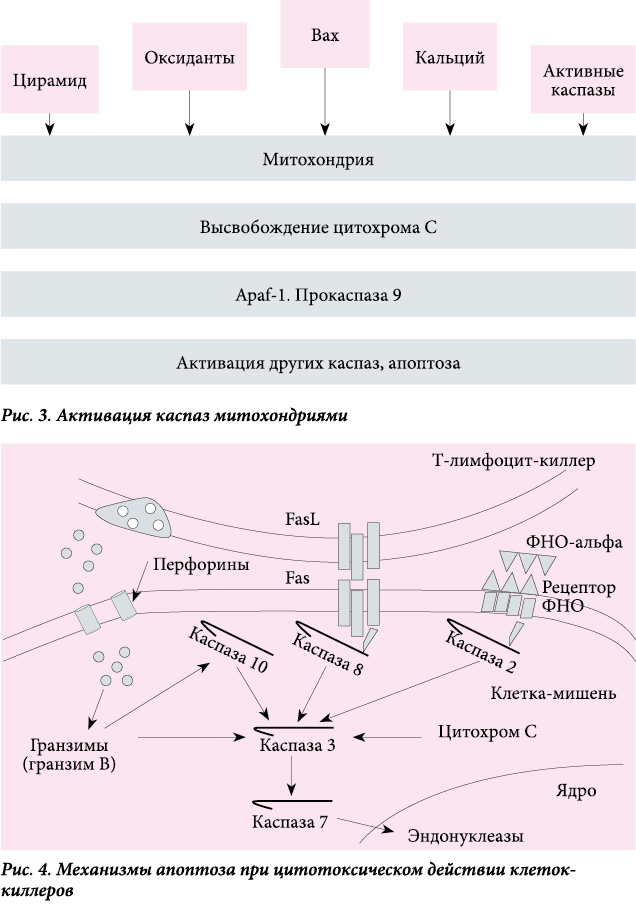
Рецепторный путь гибели клеток включается при связывании лигандов с мембранным рецептором клетки.
При связывании Fas-рецептора с Fas-лигандом включается механизм апоптоза. При этом мембраносвязываемый FasL включает сигнал апоптоза при прямом контакте клетки с клеткой, тогда как растворимая форма FasL ответственна за уничтожение клеток по типу аутокринной гибели или паракринной смерти близлежащей клетки.
Митохондриальный и рецепторный пути апоптоза активируют инициаторные каспазы. Следующий этап развития апоптоза является общим для двух указанных вариантов. Инициаторные каспазы активируют исполнительные каспазы 3, 6, 7, главной из которых является каспаза 3 [5, 9]. Мишенями исполнительных каспаз служат многочисленные белки, значительная часть которых локализуется в ядре. Расщепление молекул-мишеней определяет весь спектр проявлений апоптоза. Одна из главных мишеней каспазы 3 – эндонуклеаза CAD осуществляет дегенерацию ДНК, воздействуя на доступные участки молекулы, расположенные между нуклеосомами. Расщепление других мишеней каспаз обусловливают нарушения клеточного цикла, адгезии, клеточной морфологии и др.
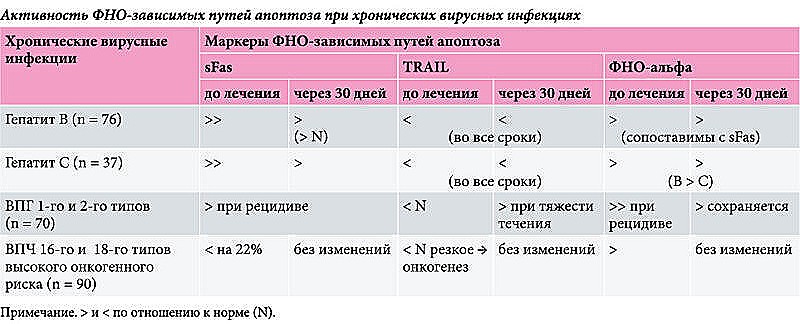
Особого внимания заслуживают механизмы апоптоза при цитотоксическом воздействии клеток-киллеров (рис. 4). Специфические Т-лимфоциты-киллеры (CD8+Т-лимфоциты) осуществляют свои киллерные функции по-разному в зависимости от наличия:
- на клетке-мишени рецепторов апоптоза, не требующих секреции литических ферментов клетками-киллерами (несекреторный лизис);
- секреторного лизиса, приводящего к запуску апоптоза под влиянием литических ферментов, вырабатываемых клетками-киллерами.
Кроме того, гибель клеток-мишеней, покрытых антителами, происходит при активации антителозависимой клеточной цитотоксичности за счет связывания антител с Fc-рецептором на клетках-киллерах (CD16) и выделения протеолитических ферментов. Гранзимы, в частности гранзим В, активируют каспазу 10. Взаимодействие Fas и FasL активирует каспазу 8, взаимодействие ФНО-альфа и рецептора ФНО-альфа – каспазу 2. Все они способны активировать основную каспазу 3, которую также активирует цитохром С. Активация каспазой 3 каспазы 7, которая активирует ядерные эндонуклеазы, завершается гибелью клетки (первый путь апоптоза). Кроме того, гранзим B способен напрямую активировать каспазу 3 и каспазу 7 (второй путь апоптоза).
Начало индукции апоптоза связано с внедрением в клетку-мишень специфического [10] белка киллерных клеток – перфорина, формирующего в мембранах поры, через которые растворимые гранзимы – сериновые протеазы клеток-киллеров попадают в клетки-мишени. Здесь они активируют целую серию цистеиновых протеаз-каспаз, включающих механизм апоптоза, который завершается активацией эндонуклеаз и фрагментацией ядра. Данный механизм разрушения клеток под влиянием Т-лимфоцитов-киллеров считается классическим при развитии реакции отторжения аллогенных трансплантатов аутоиммунной патологии, разрушении вирусинфицированных и опухолевых клеток [11]. Одновременно со специфическим механизмом киллинга Т-киллер включает и неспецифический за счет связывания свободного Fas-лиганда с Fas-рецептором клетки-мишени, что также приводит к активации цистеиновых протеаз и включению механизма апоптоза.
Апоптотические клетки и их фрагменты быстро элиминируются путем фагоцитоза, чему способствуют нарушение асимметричности мембраны и фосфатидилсерин – фермент, который в норме находится на внутренней поверхности мембраны, а при апоптозе оказывается на ее поверхности. Он распознается молекулой CD14MФ и способствует фагоцитозу клетки, на которой экспрессируется. На поверхности апоптотической клетки появляется тромбоспондин, распознаваемый молекулами адгезии – интегрином альфа-V и бета-3, CD36, через их сигналы он передается внутрь фагоцитирующей клетки и активирует ее метаболизм [9, 12].
Значение апоптоза для обеспечения нормального функционирования организма
Роль апоптоза в жизнедеятельности организма велика и сопоставима с ролью процессов пролиферации и дифференцировки клеток [12], особенно в период онтогенеза при одновременной массовой гибели клеток. У взрослых апоптоз играет ключевую роль в поддержании клеточного гомеостаза, при значительном обороте клеток, межклеточном взаимодействии и селекции клеток, прежде всего при гемопоэзе, воспалении и иммунном ответе.
При дефиците факторов выживания (Т- и B-лимфоциты, ИЛ-17, NK-клетки, ИЛ-15) избыточные клетки удаляются с помощью механизма апоптоза. Для предотвращения апоптоза достаточно индукции или поддержания уровня экспрессии антиапоптотических факторов (Bcl-2, Bcl-x1). Апоптозу подвергаются лимфоциты с нарушением перестройки антиген-распознающих рецепторов и сорецепторов, при дифференцировке субпопуляции Т-клеток [12]. При иммунопатологии и положительной селекции Т-лимфоцитов поддерживаются те клоны Т-лимфоцитов, которые распознают пептиды в составе аутологичных молекул главного комплекса гистосовместимости (ГКГ). Поддержка состоит в индукции экспрессии антиапоптотического фактора Bcl-2. В отсутствие сигналов распознавания ГКГ-клетка подвергается апоптозу [13].
На этапе отрицательной селекции элиминируются клетки, распознающие аутологические пептиды в составе аутологичных молекул ГКГ с высоким сродством. В этом случае, а также при селекции В-лимфоцитов через антиген-распознающий рецептор (TCR в Т-клетках и BCR в В-клетках) в клетки поступает сигнал, приводящий к включению апоптоза для предотвращения развития аутоиммунного процесса [5].
Апоптозу принадлежит важная роль в завершении иммунного ответа. Это проявляется в раннем развитии эффекторных клеток в соответствии с генетической программой (7–10 суток). Апоптоз также является инструментом чрезвычайной стимуляции клеток при воздействии антигена или ИЛ-2 на предварительно стимулированные клетки. Вместо дополнительной активации включается их апоптоз.
Апоптоз лежит в основе контактного цитолиза клеток-мишеней, обусловленных действием NK-клеток или цитотоксических Т-лимфоцитов. При этом индукция цитолиза может быть вызвана как инъекцией в клетку-мишень гранзима B, так и воздействием на рецепторы апоптоза.
Таким образом, роль апоптоза в иммунной системе состоит в контроле численности клеток клонального состава популяции лимфоцитов, а также в повышении сродства В-лимфоцитов и антител к антигену, ограничении продолжительности иммунного ответа.
Клиническая значимость тестов на апоптоз
Апоптоз и некроз клеток определяют при подготовке суспензии клеток для инъекций (стволовые клетки и клетки, используемые для адаптивной цитотерапии). Оценка различных типов гибели клеток используется при тестировании лекарственных препаратов, в частности цитостатиков в онкологии.
Апоптоз лимфоидных клеток изучают при анализе механизмов иммунодефицитных состояний с оценкой выраженности индукции апоптоза при активации лимфоцитов.
Спектр биологических свойств интерферона 1-го типа
Противовирусная защита организма интерфероном (ИФН) альфа-2 осуществляется за счет синтеза интерфероновых белков самим организмом в ответ на инфекцию, что обеспечивает перспективность и физиологическую значимость ИФН [14]. Доказано наличие его противовирусного действия, а именно:
- универсальность – действие на все типы вирусов в силу биологической активности;
- дистанционная внутриклеточная противовирусная активность (подавление репродукции вирусов на стадии транскрипции через активацию ферментов вместо непосредственного действия на геном вируса);
- выраженное последействие (обработанные интерфероном клетки подавляют размножение вирусов еще 24–48 часов).
ИФН-альфа, продуцируемый дендритными клетками (ДК), макрофагами и лейкоцитами, обладает выраженной противовирусной активностью. В физиологических условиях отсутствует спонтанная выработка ИФН-альфа, но сохраняется выработка его лейкоцитами при инфекциях. Клетки без внедрения вируса нечувствительны и ИФН-альфа не вырабатывают.
Первичная выработка ИФН начинается сразу после проникновения вируса в клетку через входные ворота (носоглотка, глаза, кожа). Развитие инфекции зависит от эффективности противовирусного местного иммунитета. Образованный этими клетками ИФН-альфа не обеспечивает резистентности самих клеток-продуцентов, их определенное количество погибает. Одновременно окружающие клетки под влиянием ИФН приобретают резистентность к вирусам уже через 15 минут после проникновения возбудителя.
Защитное действие ИФН-альфа, выработанного инфицированной клеткой (ДК, лейкоциты), или рекомбинантного ИФН-альфа (ВИФЕРОН) после поступления в кровь начинается со связывания со специфическими рецепторами поверхностной клеточной мембраны неповрежденных клеток, по структуре сходных с иммуноглобулином. Количество интерфероновых рецепторов на разных клетках различно, что обусловливает неодинаковую чувствительность ИФН-альфа к тканям. Сами рецепторы для ИФН-альфа отличаются друг от друга, поскольку гены, детерминирующие их синтез, локализованы в разных хромосомах: ИФН-гамма – в 18-й хромосоме, ИФН-альфа/бета – в 21-й хромосоме.
После связывания фероновыми рецепторами на внешней поверхности мембраны ИФН-альфа погружается внутрь клетки-мишени, и эта связь разрывается. Рецептор вновь возвращается на поверхность клетки, а ИФН-альфа внутри клетки через цепь сигнальных механизмов активирует транскрипцию генов, кодирующих выработку ферментов, приводящих к дегенерации чужеродной генетической информации (вирусной, геномной и матричной ДНК). Противовирусное действие ИФН-альфа обеспечивается блоком репликации вирусов на расстоянии через ферменты.
К основным функциям ИФН-альфа относятся подавление репликации вирусов за счет экспрессии противовирусных белков (Мх, GAS, PKR и др.), повышение экспрессии молекул ГКГ первого класса, усиление функции антигенпредставляющих клеток, усиление активности естественных киллеров и индукции Th1-ответа, а также индукция апоптоза.
Вирусы служат индукторами, запускающими ИФН-альфа, они чувствительны к противовирусному действию ИФН-альфа, которое реализуется через общий трансмембранный интерфероновый рецептор IFNAR-1 или IFNAR-2. Действие ИФН-альфа заключается в активации транскрипции генов ряда клеточных белков и осуществляется посредством системы межбелковых взаимодействий JAK – STAT. Сигнальный путь, инициируемый при связывании ИФН-альфа с рецептором на поверхности клетки, далее через цепь внутриклеточных механизмов активирует транскрипцию генов, индуцируемых ИФН. Показано, что при активации рецепторов к ИФН помимо активации системы JAK – STAT запускаются так называемые альтернативные сигнальные пути, определяющие разные типы биологического действия ИФН-альфа. Одновременно с индукцией транскрипции генов ряда противовирусных белков инфицированной клетки ИФН-альфа индуцирует транскрипцию генов белков – медиаторов апоптоза, таких как:
- лиганд внутриклеточного домена смерти (death domen, DD) TRAIL;
- проапоптотические белки Bak, Bax;
- протеинкиназы RKR, которые индуцируют апоптоз с помощью FADD (Fas-associated death domen protein), опосредованной активации каспазы 8;
- активирующие факторы IRF;
- ген промиелоцитарной лейкемии PML.
Наряду с индукцией синтеза противовирусных белков ИФН-альфа активирует апоптоз инфицированных вирусом клеток, что обеспечивает их деструкцию, снижает размножение вируса и распространение инфекции. Помимо универсального противовирусного и иммуномодулирующего действия ИФН 1-го типа оказывает дополнительное индуцирующее (стимулирующее) или ингибирующее (угнетающее) разнонаправленное действие на различные звенья эпителиальных и иммунных клеток, которые могут опосредовать развитие патологических состояний и тканевых повреждений (в том числе через механизмы апоптоза).
Доказано наличие прямого усиливающего эффекта ИФН-альфа на продукцию ИЛ-10 – противовоспалительного цитокина, который вторично прямо или опосредованно блокирует провоспалительные ИЛ-1, ИЛ-2, ИЛ-17, ИФН-гамма и колониестимулирующий фактор, снижая уровень Т-клеток, ИЛ-12 и ИФН-гамма, приводящих к иммуносупрессии. Прямое повышение уровня ИЛ-27 под воздействием ИФН-альфа приводит к продукции ИЛ-17 и впоследствии к антибактериальному ответу.
ИФН 1-го типа также оказывает прямой модулирующий (стимулирующий или ингибирующий) эффект в отношении продукции антител и уровня CD8+ цитотоксических Т-лимфоцитов в зависимости от состояния иммунной системы.
Кроме того, доказано прямое влияние ИФН 1-го типа на механизм запуска апоптоза через модуляцию активирующих или ингибирующих его генов.
ИФН-альфа вызывает апоптоз клетки в стадии транскрипции генов белков – медиаторов апоптоза. ИФН-альфа может также запускать механизм апоптоза прямо через индукцию двух основных мембранных клеточных рецепторов, принимающих сигналы к развитию апоптоза: FAS (CD95-APO 1) и рецептора ФНО 1-го типа TRAIL с их соответствующими лигандами. Эта пара запускает апоптоз – программируемую гибель клеток-мишеней.
Обобщение биологических эффектов ИФН-альфа/бета через механизм апоптоза представлено на рис. 5.
![]()
Запускаемый ИФН 1-го типа апоптоз эпителиальных клеток респираторного тракта, гепатоцитов, иммунных Т-клеток, высокостимулированного воспаления может приводить к разрушению тканевых клеток или иммуносупрессии.
Подходы к оценке апоптоза при клинико-иммунологических исследованиях
Определение апоптоза основано на регистрации феноменов, лежащих в его основе. Речь идет о формировании разрывов ДНК, развивающихся вследствие этого деградации и потере клеткой части ДНК, асимметрии мембраны с экспрессией на поверхности необычных молекул, изменении морфологии клетки.
В настоящее время для регистрации апоптоза, в том числе при работе с лимфоцитами, широко применяются методы проточной цитофлуориметрии. При скрининговых исследованиях используется метод, основанный на выявлении гиподиплоидных клеток (утрата части хроматида). Чрезвычайно удобным и информативным считается метод, при котором регистрируется ранний признак апоптоза – экспрессия на поверхности клеток фосфатидилсерина. Для обнаружения экспрессии фосфатидилсерина используют конъюгат аннексина V, который обладает сродством к фосфорилсерину, с флуоресцеинизотиоцианатом. Преимуществом метода является возможность регистрации апоптоза на ранних этапах его развития, а также надежная дифференциация апоптоза от некроза [3, 9].
Оценка экспрессии на поверхности лимфоцитов Fas-рецептора (CD95) и в митохондриях протоонкогена Bcd-2 проводится для выявления высокого риска развития апоптоза. Метод наиболее эффективен при взаимодействии с Fas-рецептором клетки-мишени с помощью проточной цитофлуориметрии. Использование этих комбинаций позволяет оперативно оценить процент лимфоцитов, подвергшихся апоптозу. Еще один важный аспект для оценки апоптоза в клинической практике – использование индукции апоптоза для получения расширенной информации [8, 12].
Помимо подавления трансляции вирусного генома ИФН-альфа индуцирует клеточный апоптоз, способствуя элиминации инфекционного агента за счет гибели клетки.
Регулирование механизмов апоптоза препаратами интерферона в клинической практике при вирусных инфекциях
Вирусы могут оказывать иммуносупрессивное действие, направленное как на нарушение синтеза цитокинов, так и на снижение апоптотической активности клеток, которые персистируют в организме даже в отсутствие активной репликации вируса за счет инфицирования геномом. Определение активности ФНО-зависимых путей апоптоза – классических путей запуска программируемой гибели клеток является адекватной моделью для поиска способов регуляции апоптоза и обоснования методов его коррекции в клинической практике, в том числе с использованием интерфероновых препаратов [5, 12, 15].
Активность ФНО-зависимых путей апоптоза при хронических вирусных инфекциях
При оценке активности ФНО-зависимых путей апоптоза нами был исследован уровень маркеров апоптоза в сыворотке крови пациентов с хроническими вирусными инфекциями: гепатитом B (n = 66), С (n = 37), рецидивирующей герпесвирусной инфекцией (вирус простого герпеса (ВПГ) 1-го и 2-го типов), цитомегаловирусной инфекцией (ЦМВ) (n = 70) и вирусом папилломы человека (ВПЧ) высокого онкогенного риска (n = 94) [16, 17]. При формировании групп учитывали критерии отбора пациентов для обследования, верификацию диагноза на основании клинических и лабораторных данных, тяжесть клинического течения.
В качестве маркеров апоптоза использовали рецепторы, связывание которых с лигандами индуцирует апоптоз в клетке-мишени, являющейся носителем сывороточного растворимого Fas-рецептора (sFas), TRAIL, ФНО-альфа в динамике методом твердофазного иммуноферментного анализа с использованием стандартных тест-систем [7]:
- р1ФНО (рецептор 1-го типа для ФНО) и семейство родственных ему TRAIL (TRAIL – ФНО-опосредованный лиганд, индуцирующий ФНО-запускающий апоптоз, родственный ФНО) [18];
- CD95 (APO-1) – Fas-рецептор и FasL;
- ФНО [18].
Обследование проводили от начала лечения или с момента рецидива инфекции. Через месяц к стандартной терапии добавляли ациклические нуклеозиды и человеческий рекомбинантный препарат ИФН 1-го типа прямого противовирусного и иммуномодулирующего действия в комплексе с высокоактивными антиоксидантами витаминами E и C (ВИФЕРОН) [16, 17]. Результаты исследования активности ФНО-хронических вирусных инфекций представлены в таблице [17].
При поступлении в стационар у больных вирусным гепатитом B отмечалось существенное повышение концентрации sFas. На 30-е сутки после поступления показатели sFas достоверно снижались, но не приходили в норму. Показатели изменения концентрации sFas при гепатите C были аналогичны показателям при вирусе гепатита B. Даже через 30 дней этот показатель был самый высокий в группах.
При изучении концентрации ФНО в сыворотке крови пациентов с гепатитами зафиксированы данные, сопоставимые с показателями sFas. Наиболее высокие концентрации ФНО наблюдались при гепатите B и достоверно отличались от аналогичных показателей при гепатите С. В отличие от показателей Fas и ФНО показатели TRAIL демонстрировали противоположную тенденцию. Во всех периодах обследования отмечалось достоверное снижение концентрации исследуемого фактора, самые низкие значения наблюдались при гепатите С. При оценке активности апоптоза у больных ВПГ 1-го и 2-го типов отмечалось повышение sFas в момент рецидива в зависимости от степени тяжести: при легкой степени повышение составило 42%, при средней степени – 35%, при тяжелой – 26%. Через 30 суток с момента лечения изменений не зарегистрировано. Снижение показателя sFas свидетельствует об уменьшении запуска клеткой апоптоза.
Тенденция к снижению TRAIL в сыворотке крови отмечалась у пациентов с более тяжелым течением инфекционного процесса. Концентрации TRAIL оказались достоверно ниже в группе доноров (р
Заключение
В настоящее время доказано участие системы интерферона в процессе апоптоза. Цикл противовирусного действия ИФН-альфа/бета (1-го типа) сопровождается такими процессами, как иммуномодуляция, индукция апоптоза, блокада пролиферации клеток, регулируемых интерферонами. Это говорит о разнонаправленном действии ИФН-альфа/бета. Поскольку апоптоз – физиологический процесс, в организме имеется множество специфических факторов ингибирующего или регуляторного действия, запускающих процесс апоптоза клетки. К факторам регуляторного действия относится большая группа белков – цитокинов, регулирующих пролиферацию и дифференцировку клеток при связывании со специфическими рецепторами на клетках-мишенях и при патологических ситуациях. Доказано также участие белков интерфероновой системы в процессе регуляции апоптоза при разных патологиях, что особенно важно при вирусных инфекциях.
Условно весь процесс апоптоза может быть разделен на две фазы – формирование и проведение апоптотических сигналов и демонтаж клеточных структур с участием особых протеаз (каспаз). Принято выделять два взаимосвязанных механизма активации каспаз – рецепторный (на поверхности неповрежденных клеток) и митохондриальный (для патологически измененных клеток). При исследовании апоптотических свойств ИФН-альфа/бета (1-го типа) доказана его способность вызывать ингибирование апоптогенного сигнала для моноцитов (один из механизмов гибели клеток). ВИФЕРОН – препарат ИФН 1-го типа также является ингибитором апоптогенного сигнала для моноцитов. ИФН альфа-2b вызывает апоптоз клеток в стадии транскрипции через активацию генных белков.
Особо следует отметить роль белка Вid – связующего звена между рецепторным и митохондриальным механизмами активации каспаз.
Таким образом, актуальность изучения процесса апоптоза с выявлением механизмов нарушения его регуляции, сопровождаемых конкретными заболеваниями, позволяет определять этиологию и патогенез данных заболеваний и возможность коррекции нарушения регуляции программируемой гибели клетки. Идентификация и регуляция морфологических и биохимических маркеров апоптоза, включая апоптоз специфических генов, важны для понимания механизма патогенеза заболевания, дифференциальной диагностики и разработки принципиально новых направлений терапии.
Включение в схему комплексного лечения препарата ВИФЕРОН подтверждает его эффективность в коррекции уровня апоптоза в зависимости от клинико-иммунологической патологии ФНО-зависимых путей апоптоза при хронических вирусных инфекциях, в том числе высокого онкогенного риска.
Литература
- Манских В.К. Пути гибели клетки и их биологическое значение // Цитология. 2007. Т. 49. № 11. С. 909–915.
- Kerr J.F., Wyllie A.H., Currie A.R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics // Br. J. Cancer. 1972. Vol. 26. № 4. P. 239–257.
- Варга О.Ю., Рябков В А. Апоптоз: понятие, механизмы реализации, значение // Экология человека. 2006. № 7. С. 28–32.
- Фомченко Н.Е., Воропаев Е.В. Биологические аспекты апоптоза (обзор литературы) // Проблемы здоровья и экологии. 2013. № 1 (35). С. 39–45.
- Григорьев М.Ю. Апоптоз в норме и патологии // Медицинский академический журнал. 2003. Т. 3. № 3. С. 3–11.
- Москалева Е.Ю., Северин С.Е. Возможные механизмы адаптации клетки к повреждениям, индуцирующим программную гибель. Связь с патологией // Патологическая физиология и экспериментальная терапия. 2006.№ 2. С. 2–16.
- Holoch P.A., Griffith T.S. TNF-related apoptosis-inducing ligand (TRAIL): a new path to anti-cancer therapies // Eur. J.Pharmacol. 2009. Vol. 625. № 1–3. P. 63–72.
- Червякова Н.В. Fas/Fas-лиганд: маркеры апоптоза // Лаборатория. 2004. № 2. С. 7–9.
- Хаитов Р.М., Пинегин Б.В., Ярилин А.А. Руководство по клинической иммунологии. Диагностика заболеваний иммунной системы. М.: ГЭОТАР-Медиа, 2009.
- Crouse J., Bedenikovic G., Wiesel M. et al. Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1 // Immunity. 2014. Vol. 40. № 6. P. 961–973.
- Crouse J., Xu H.C., Lang P.A., Oxenius A. NK cells regulating T cell responses: mechanisms and outcome // Trends Immunol. 2015. Vol. 36. № 1. P. 49–58.
- Ярилин А.А., Никонова Н.Ф., Ярилина А.А. и др. Апоптоз, роль в патологии и значимость его оценки при клинико-иммунологическом обследовании больных (обзор) //Медицинская иммунология. 2000. Т. 2. № 1. С. 7–16.
- MacMicking J.D. Interferon-inducible effector mechanisms in cell-autonomous immunity // Nat. Rev. Immunol. 2012. Vol. 12. № 5. P. 367–382.
- De Weerd N.A., Samarajiwa S.A., Hertzog P.J. Type I interferon receptors: biochemistry and biological functions // J. Biol. Chem. 2007. Vol. 282. № 28. P. 20053–20057.
- Исаков В.А., Архипова Е.И., Исаков Д.В. Герпес-вирусные инфекции человека. Руководство для врачей. СПб.: СпецЛит, 2013.
- Соколова Т.М., Успелевский Г.П., Колодяжная Л.В. и др. Эффекты индукторов интерферона на экспрессию генов-регуляторов апоптоза в лимфоцитах человека // Цитокины и воспаление. 2011. Т. 10. № 2. С. 75–80.
- Хараева З.Ф., Иванова М.Р., Кузьмицкая Е.Ф. и др. Показатели активности TNF-зависимых путей апоптоза при хронических вирусных инфекциях с высоким онкогенным риском // Цитокины и воспаление. 2010. Т. 9.№ 4. С. 127–128.
- Bernardo A.R., Cosgaya J.M., Aranda A., Jimenez-Lara A.M. Synergy between RA and TLR3 promotes type I IFN-dependent apoptosis through upregulation of TRAIL pathway in breast cancer cells // Cell Death Dis. 2013. Vol. 4. IDe479.
- Meylan E., Tschopp J. Toll-like receptors and RNA helicases: two parallel ways to trigger antiviral responses // Mol. Cell. 2006. Vol. 22. № 5. P. 561–569.
- Rajasuriar R., Khoury G., Kamarulzaman A. et al. Persistent immune activation in chronic HIV infection: do any interventions work? // AIDS. 2013. Vol. 27. № 8. P. 1199–1208.
- Chen L., Borozan I., Feld J. et al. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection // Gastroenterology. 2005. Vol. 128. № 5. P. 1437–1444.
- Davidson S., Crotta S., McCabe T.M., Wack A. Pathogenic potential of interferon ?? in acute influenza infection // Nat. Commun. 2014. Vol. 5. ID3864.
- Davidson S., Maini M.K., Wack A. Disease-promoting effects of type I interferons in viral, bacterial, and coinfections // J. Interferon Cytokine Res. 2015. Vol. 35. № 4.
P. 252–264.
Mechanisms and Efficiency of Apoptosis Regulation by Viferon
ООО ФеронV.V. Malinovskaya1, V.S. Suskova2, Ye.N. Vyzhlova1, A.N. Shuvalov1, S.I. Suskov3
1 N.F. Gamaleya Federal Research Center for Epidemiology and Microbiology
2 ‘Diagnostics’ Medical Center (Moscow)
3 ‘SM-KLINIKA’ Group of Companies (Moscow)
Contact person: Valentina Vasilyevna Malinovskaya, eym@viferon.su
The end of the XX century was commemorated by achievements in biology and medicine, including theoretical immunology, which allowed taking a fresh look at the etiopathogenesis of a number of diseases. Specifically, we are talking about changes concerning the concepts of innate immunity, the discovery of the interferon system, the identifi cation of immunoregulatory cells and lymphocyte subpopulations that synthesize oppositely acting cytokines, as well as their role in autoimmune pathology.
Key words: innate immunity, immunoregulatory cells, apoptosis, interferon, Viferon
Впервые опубликовано в журнале «Эффективная фармакотерапия. Эпидемиология и инфекции», 2018, № 15/1, С. 22-31



